elocation-id: e3956
The sweet orange is grown in several states of Mexico. Several research centers have citrus germplasm banks, including sweet oranges for genetic improvement. This work aimed to evaluate genetic diversity and perform a phylogenetic analysis based on the internal transcribed spacer (ITS) region of six sweet orange genotypes grown in San Luis Potosí, Mexico. In 2023, leaves of six varieties of sweet orange, known in the region as Sangre de Toro and Valencia, were collected. Genomic DNA was extracted, and ITS regions were amplified by PCR and sequenced. The percentage of identity and genetic distances were determined and a maximum-likelihood phylogenetic tree was generated. The results indicated that the intraspecific variability among C. sinensis genotypes in the ITS region is low to moderate (0-0.07). Particularly, among the population of Citrus sinensis (Sangre de Toro and Valencia varieties) from the regions of Axtla de Terrazas, Chimimexco-Tampacán and El Frijolillo-San Martín in San Luis Potosí, no differences were detected in their ITS regions. It is important to note that the number of samples analyzed was limited; therefore, it is necessary to evaluate a larger set of samples and varieties to obtain more robust conclusions regarding genetic variability in Citrus sinensis in Mexico. The phylogenetic analysis grouped the genotypes of Mexico into group G1, confirming the origin of the varieties with Asian countries (China, Korea and Vietnam), with which the percentage of identity was 100%.
Citrus sinensis, genetic variability, sequencing
The sweet orange (Citrus sinensis) is one of the most consumed citrus worldwide. The fruits are highly appreciated for their organoleptic characteristics and high nutraceutical value, mainly for their high antioxidant content (Seminara et al., 2023). The genetic characterization of C. sinensis materials is important for breeding programs that seek to cross various materials to generate varieties with desirable genetic and commercial traits (Almeyda-León et al., 2023). In this sense, work has been carried out to determine the genetic variability and phylogenetic analyses of a wide range of C. sinensis varieties using various molecular markers, such as random amplified microsatellites (RAMs), random amplified polymorphic DNA (RAPDs), simple sequence repeats (SSRs) and start codon targeted (SCoT).
Abbaszadeh et al. (2023) determined low to moderate genetic diversity in 29 sweet orange genotypes from Iran using the SCoT molecular marker. The population showed a genetic structure influenced by geographical distance and climatic variables (temperature and precipitation). In another study, Shahnazari et al. (2022) conducted a genetic diversity analysis of 80 sweet orange trees of different varieties from Iran using SSR markers, finding high genetic variability that supports the use of these markers for identifying and classifying varieties. On the other hand, Tuwo et al. (2023) employed RAPD markers to analyze the genetic diversity of citrus species in five production centers in Indonesia, finding a high level of polymorphisms and close relationships among some genotypes.
In Mexico, Almeyda-León et al. (2022) carried out the genetic characterization of a population of 33 genotypes of sweet orange (C. sinensis) from the germplasm bank of the General Terán Experimental Field of the National Institute of Forestry, Agricultural and Livestock Research (INIFAP, by its Spanish acronym) in Nuevo León, Mexico, using RAM markers. All of these molecular markers produce binary matrices of the presence or absence of fragments to analyze genetic diversity, but their development can be laborious and error-prone. Alternatively, procedures such as DNA barcoding can be used, thus increasing the accuracy of varietal characterization.
In plants, nuclear regions (ITS, ITS1 and ITS2) and chloroplast regions (matK, rbcL, trnH-psbA, rpoB, rpoC1, trnL-trnF and psbK-psbI) have been used (Letsiou et al., 2024). These markers have been used for phylogenetic analyses with several citrus species, including C. sinensis (Li et al., 2010; Kyndt et al., 2010; Penjor et al., 2013; Hynniewta et al., 2014; Ito et al., 2014; Jin et al., 2018; Liu et al., 2021; Abbaszadeh et al., 2023). However, most studies are focused on Asian countries. In Latin America, there are no studies that have used the ITS region in C. sinensis to determine its diversity.
The objective of this work was to assess genetic diversity and perform a phylogenetic analysis based on the ITS region of six C. sinensis genotypes grown in San Luis Potosí, Mexico.
Young, healthy-looking leaves were collected from six sweet orange trees (Table 1). The leaves were wrapped in paper bags and placed in thermal coolers with refrigerant gels to be sent to the Biotechnology Laboratory of INIFAP’s Tecomán Experimental Field in Colima, where they were immediately processed.
The method used was that described by Bermúdez-Guzmán et al. (2016) with some modifications. Plant tissue (approximately 200 mg) was macerated in mortars and homogenized in 2 ml of CTAB buffer preheated at 65 °C for 60 min and treated with RNase A, followed by phase separation with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitation of DNA with cold isopropanol and sodium acetate. The DNA pellet was washed with 70% ethanol, dried at room temperature, and resuspended in water.
Quantification and purity were determined from 2 μl of each sample, measuring the 260/280 and 260/230 nm absorbance ratios using a NanoDrop 2000 spectrophotometer (Thermo Scientific). DNA integrity was verified by 1% agarose gel electrophoresis in 1X TAE buffer. Nucleic acid staining was performed with GelRed (Biotium) and visualized on a gel documentation system (UVP).
The oligonucleotides used were those described by White et al. (1990), modified by Viglietti et al. (2019): ITS4: 5’-TCCTCYRMTTAKYGATATGC-3’ and ITS1: 5’-TCCGTWRGTGAACCWGCGG-3’. PCR reactions were performed in a volume of 50 μl using REDTaq® ReadyMixTM PCR Reaction Mix (Sigma Aldrich), according to the manufacturer’s instructions.
The annealing temperature was standardized using a gradient: 50, 51, 52, 53, 54 and 55 °C in a VeritiTM thermal cycler (Applied BiosystemsTM). For all cases, the amplification conditions were 5 min at 95 °C, followed by 35 cycles of 1 min at 95 °C, 30 s at 50-55 °C, and 1 min at 72 °C, and a final extension of 10 min at 72 °C. PCR products between 600 and 700 bp were purified with the Wizard® SV Gel and PCR Clean-Up System (Promega) and sent for sequencing to the National Laboratory of Agricultural, Medical, and Environmental Biotechnology (Lanbama, by its Spanish acronym).
The sequences were edited, a BLAST analysis was performed in the NCBI database, and 28 sequences of the ITS regions from C. sinensis were downloaded to generate a local database. As an external group, an ITS sequence from Ruta graveolens was included, and a multiple sequence alignment (MSA) was performed using the ClustalW algorithm.
To analyze genetic diversity, sequence-comparison matrices were generated based on the MSA. The distance matrix was calculated as the Jukes-Cantor corrected distance between two sequences. For the phylogenetic analysis, the MSA was analyzed with the ‘model test’ tool. A phylogenetic tree was constructed using the UPGMA construction method with the GTR + G + T nucleotide substitution model and a Bootstrap analysis with 1 000 replicates. All the aforementioned bioinformatic analyses were carried out using a valid license of the CLC Main Workbench software, version 24.0.2 (Qiagen).
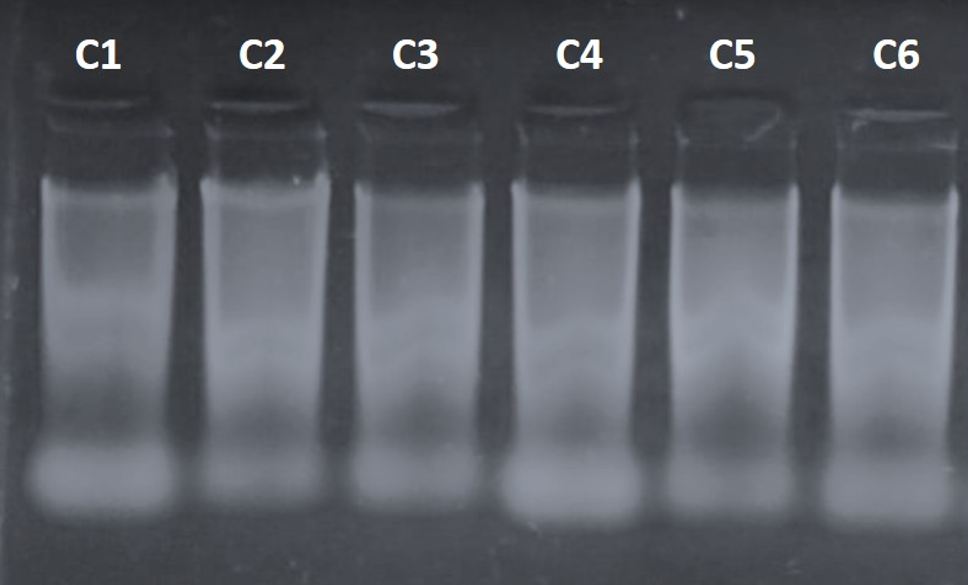
The average purity indices were 1.8 (A260/280) and 1.79 (A260/230). Figure 1 presents the agarose gel electrophoresis of the extracted genomic DNA, which shows partial degradation of the samples, likely due to the time elapsed since transport from San Luis Potosí to Colima. Nonetheless, in the upper part of the gel, high-integrity DNA bands were observed, sufficient for PCR amplification of the ITS region and for obtaining sequences.
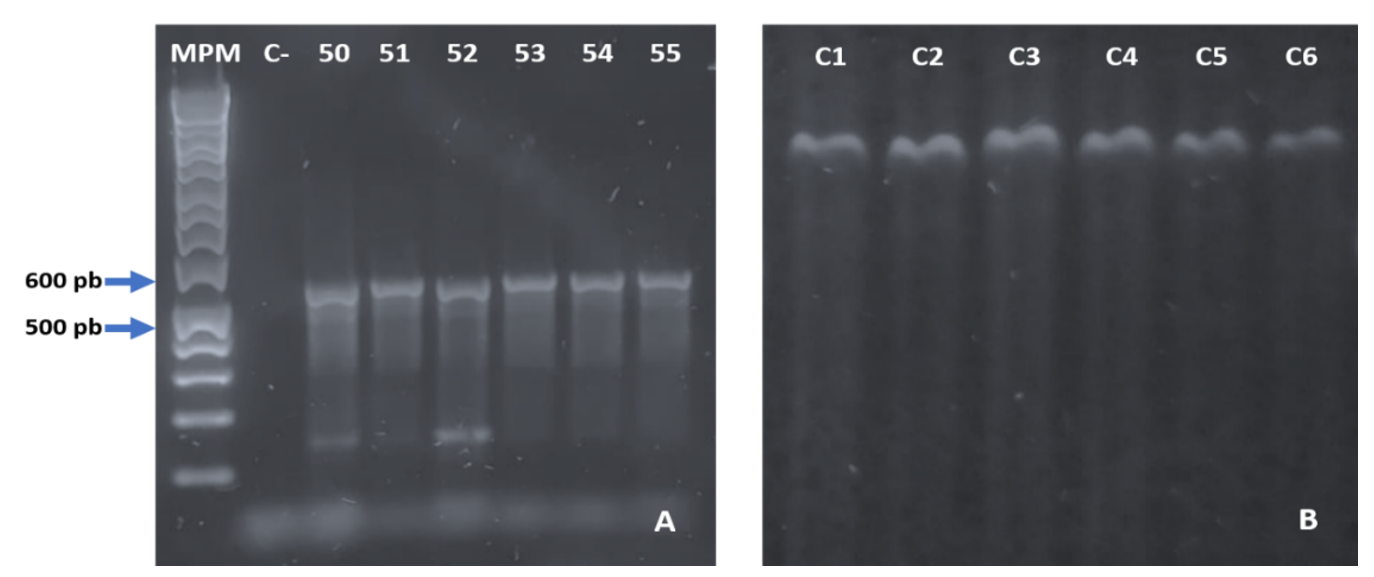
Gradient PCR amplification to assess the effect of temperatures of 50-55 °C is shown in Figure 2A. Temperatures of 53 to 55 °C showed unique bands, and the temperature of 54 °C was selected to amplify the six orange tree samples by PCR (Figure 2B).
In the phylogenetic tree of several C. sinensis genotypes from different regions of the world, it is observed that the 28 genotypes were distributed in five groups (G1-G5) (Figure 3). The Mexican genotypes formed a group with others from Korea, China and Vietnam (G1), with some of which they share 100% identity (JN681150, MG702211, MG702223, MF15502 and ON479687), constituting the most closely related materials.
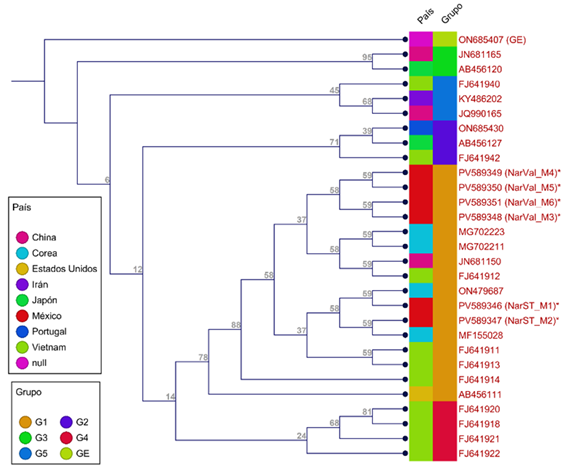
Since increasing geographic distance often increases genetic distance (Abbaszadeh et al., 2023), in this study, the genetic variability in the ITS region among the six genotypes of C. sinensis was 0, with homologous sequences, probably due to the proximity of the sampling sites and the limited number of samples analyzed. This indicates high conservation between the Sangre de Toro and Valencia varieties in this region. The genotypes from Portugal, Japan and Vietnam, which form group G5 (Figure 3), showed greater genetic distance, with values of 94.1-95.71% of identity and a distance coefficient of 0.04-0.06.
The ITS sequence is widely used to identify plant species and conduct phylogenetic studies (Alvarez and Wendel, 2003; Graper et al., 2021). In citrus, it has been successfully used in species such as C. sinensis, C. limon and C. reticulata to evaluate genetic variability and phylogenetic relationships (Hynniewta et al., 2014). In this study, the genetic distance between all genotypes of C. sinensis (28 sequences) was low to moderate (0-0.07), indicating high conservation in the ITS region, whereas for Ruta graveolens, the values were 0.24-0.29. Similar results were obtained by Liu et al. (2021) in 22 samples of C. reticulata ‘Chachi’, where the ITS2 region showed a 100% homology, indicating high genetic stability in this region.
On the other hand, the genotypes of C. sinensis from Vietnam showed greater intraspecific variability (distances of 0.02-0.06) and were grouped into G1, G2, G4, and G5. Materials from China were in G1, G3, and G5, whereas those from Korea and Mexico clustered in G1. This diversity can be attributed to different varieties, climatic regions and cultivation and breeding practices (Abbaszadeh et al., 2023; Seminara et al., 2023). In contrast to these results, Almeyda-León et al. (2022) evaluated 33 varieties of sweet orange trees using RAMs, achieving a high percentage of polymorphisms and differentiating all the materials evaluated, which indicates a broad genetic base in their germplasm.
In other studies, using RAPDs, Tuwo et al. (2023) identified five groups with 175 citrus genotypes using cluster analysis with a similarity coefficient of 77%. On the other hand, SSR markers have also been used to identify hybrids for genetic improvement in citrus (Bermúdez-Guzmán et al., 2017; Gallego-Colonia et al., 2017; Sharafi et al., 2017; Carrillo-Medrano et al., 2018). This suggests that the use of molecular markers based on electrophoretic profiles, such as RAPDs, RAMs, SSRs and SCoT, among others, is still very useful for characterizing genetic variability in C. sinensis and other citrus species (Shahnazari et al., 2022; Abbaszadeh et al., 2023).
The use of these molecular markers for plant characterization dates back to the 90s (Williams et al., 1990; Morgante and Olivieri, 1993), but they are still widely used today because they are highly versatile, as they explore the entire genome. This study evaluated a small region of a genome, ITS, with a length of just over 550 bp, which represents a tiny part of the entire genome. For this reason, to make the diversity analysis more robust, it is advisable to use more than one region, such as nuclear regions (ITS, ITS1 and ITS2) or chloroplast regions (matK, rbcL, trnH-psbA, rpoB, rpoC1, trnL-trnF, psbK-psbI) (Letsiou et al., 2024).
On the other hand, it is important to consider that the costs of massive sequencing (NGS) have decreased significantly, so in the near future, it is desirable to migrate to this type of marker. With this technology, a large amount of information can be obtained on genetic variation, such as single-nucleotide polymorphisms (SNPs), insertion-deletions (InDels), structural variation (SV) and copy number variation (CNV), among others (Wenger et al., 2019; Ahsan et al., 2021).
The ITS region can be used as an exploratory and comparative tool to evaluate phylogenetic relationships in the genus Citrus; however, its intraspecific resolution was limited in closely related materials, as was the case with C. sinensis genotypes. In this study, with six samples corresponding to two varieties from San Luis Potosí (SLP), a low to moderate intraspecific variability (0-0.07) was observed between various genotypes of C. sinensis from various parts of the world. These findings are specific to the samples analyzed from the SLP region. The genotype sequences showed similarities with Asian accessions (China, Korea and Vietnam), but expanding genomic coverage is required to confirm these possible origins.
We are grateful to INIFAP for financing the project entitled: ‘Search for natural citrus mutants with characteristics of agronomic, phytosanitary and industrial interest in the northeast region of Mexico’.
Almeyda-León, I. H.; Narvaéz-Rodríguez, Á. I.; Pecina-Quintero, V.; Álvarez-Ojeda, M. G. y Núñez-Colín, C. A. 2023. Caracterización y diversidad genética de cítricos del banco de germoplasma del Campo Experimental General Terán. Biotecnología y Sustentabilidad. 8(1):26-41. https://doi.org/10.57737/biotecnologiaysust.v8i1.2276.
Almeyda-León, I. H.; Pecina-Quintero, V.; Álvarez-Ojeda, M. G.; Rodríguez-Guerra, R.; Acosta-Díaz, E. y Núñez-Colín, C. A. 2022. Caracterización y diversidad genética de naranjos dulces (Citrus sinensis L.) del banco de germoplasma del Campo Experimental General Terán. Biotecnología y Sustentabilidad. 7(1):80-95. https://doi.org/10.57737/biotecnologiaysust.v7i1.1645.
Bermúdez-Guzmán, M. J.; Guzmán-Rodríguez, L. F.; García-Mariscal, K. P.; Palmeros-Suárez, P. A. y Orozco-Santos, M. 2017. Identificación de híbridos de Citrus aurantifolia×Citrus limon utilizando marcadores de secuencias simples repetidas (SSR). Revista Mexicana de Ciencias Agrícolas. 8(6):1397-1408. https://doi.org/10.29312/remexca.v8i6.309.
Gallego-Colonia, J. S.; Enríquez-Valencia, A. L.; Caicedo-Arana, Á.; Posso-Terranova, A. M. y Muñoz-Florez, J. E. 2017. Diversidad genética en patrones de cítricos mediante microsatélites amplificados al azar (RAMs). Biotecnología en el Sector Agropecuario y Agroindustrial. 15(1):85-94. https://doi.org/10.18684/BSAA(15)85-94.
Ito, T. M.; Polido, P. B.; Rampim, M. C.; Kaschuk, G. and Souza, S. G. H. 2014. Genome-wide identification and phylogenetic analysis of the AP2/ERF gene superfamily in sweet orange (Citrus sinensis). Genetics and Molecular Research: GMR. 13(3):7839-7851. https://doi.org/10.4238/2014.September.26.22.
Jin, S. B.; Lee, W. J.; Park, J. H.; Park, S. M.; Lee, D. H. and Yun, S. H. 2018. A phylogenic analysis of citrus cultivars native to jeju using chloroplast dna trnl-trnf and internal transcribed spacer region sequences. Horticultural Science and Technology. 36(4):585-597. https://doi.org/10.12972/kjhst.20180059.
Li, X.; Xie, R.; Lu, Z. and Zhou, Z. 2010. The origin of cultivated citrus as inferred from internal transcribed spacer and chloroplast DNA sequence and amplified fragment length polymorphism fingerprints. Journal of the American Society for Horticultural Science. 135(4):341-350. https://doi.org/10.21273/JASHS.135.4.341.
Liu, M.; Wang, K.; Chen, B.; Cai, Y; Li, C.; Yang, W.; Wei, M. and Zheng, G. 2021. Intraspecific DNA barcoding and variation analysis for Citri reticulatae pericarpium of Citrus reticulata ‘Chachi’. Evidence-based Complementary and Alternative Medicine. eCAM, 2021, 2609935: 1-7 pp. https://doi.org/10.1155/2021/2609935.
Wenger, A. M.; Peluso, P.; Rowell, W. J.; Chang, P. C.; Hall, R. J.; Concepcion, G. T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N. D.; Töpfer, A.; Chin, C. S.; Alonge, M.; Mahmoud, M.; Qian, Y.; Phillippy, A. M.; Schatz, M. C.; Myers, G.; Pristo, M. A. and Hunkapiller, M. W. 2019. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. Preprint, bioRxiv, 519025. https://doi.org/10.1101/519025.
White, T.; Bruns, T.; Lee, S.; Taylor, J.; Innis, M.; Gelfand, D. and Sninsky, J. J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols: A Guide to Methods and Applications. Academic Press. San Diego, California, USA. 315-322 pp. https://doi.org/10.1016/b978-0-12-372180-8.50042-1.