elocation-id: e3956
La naranja dulce se cultiva en varios estados de México. Diversos centros de investigación poseen bancos de germoplasma de cítricos, donde se incluye la naranja dulce para su mejoramiento genético. El objetivo del trabajo fue evaluar la diversidad genética y realizar un análisis filogenético basados en la región del espaciador transcrito interno (ITS) de seis genotipos de naranja dulce cultivados en San Luis Potosí, México. En 2023 se recolectaron hojas de seis variedades de naranja dulce conocidas en la región como Sangre de Toro y Valencia. Se extrajo el DNA genómico, se amplificaron regiones ITS por PCR y se secuenciaron. Se determinó el porcentaje de identidad, distancia genética y se generó un árbol filogenético de máxima verosimilitud. Los resultados indicaron que la variabilidad intraespecífica entre genotipos de C. sinensis en la región ITS es baja a moderada (0-0.07). Particularmente, entre la población de Citrus sinensis (variedades Sangre de Toro y Valencia) de las regiones de Axtla de Terrazas, Chimimexco-Tampacán y El Frijolillo-San Martín en San Luis Potosí, no se detectaron diferencias en sus regiones ITS. Es importante señalar que el número de muestras analizadas fue limitado; por lo tanto, se requiere evaluar un conjunto mayor de muestras y variedades para obtener conclusiones más robustas respecto a la variabilidad genética en Citrus sinensis en México. El análisis filogenético agrupó a los genotipos de México en el grupo G1, confirmando el origen de las variedades con países asiáticos (China, Corea y Vietnam), con los cuales el porcentaje de identidad fue del 100%.
Citrus sinensis, secuenciación, variabilidad genética
La naranja dulce (Citrus sinensis) es uno de los cítricos más consumidos a nivel mundial. Los frutos son muy apreciados por sus características organolépticas y el alto valor nutracéutico, principalmente por su alto contenido de antioxidantes (Seminara et al., 2023). La caracterización genética de los materiales de C. sinensis es importante para los programas de mejoramiento genético que buscan realizar cruzas entre diversos materiales para generar variedades con características genéticas y comerciales deseables (Almeyda-León et al., 2023). En este sentido, se han realizado trabajos para determinar la variabilidad genética y análisis filogenéticos de una amplia gama de variedades de C. sinensis utilizando diversos marcadores moleculares como random amplified microsatellite (RAM), RAPDs random amplified polymorphic (DNA), simple sequence repeat (SSR) y start codon targeted (SCoT).
Abbaszadeh et al. (2023) determinaron diversidad genética de baja a moderada en 29 genotipos de naranja dulce procedentes de Irán utilizando el marcador molecular SCoT. La población mostró una estructura genética influenciada por la distancia geográfica y variables climáticas (temperatura y precipitación). En otro trabajo, Shahnazari et al. (2022) realizaron un estudio de diversidad genética en 80 árboles de naranja dulce de diferentes variedades procedentes de Irán utilizando marcadores SSR, encontrando una alta variabilidad genética que respalda el uso de estos marcadores para la identificación y clasificación de variedades. Por otra parte, Tuwo et al. (2023) utilizaron marcadores RAPD para analizar la diversidad genética de especies de cítricos en cinco centros de producción en Indonesia, encontrando un alto nivel de polimorfismos y relaciones cercanas entre algunos genotipos.
En México, Almeyda-León et al. (2022) realizaron la caracterización genética de una población de 33 genotipos de naranja dulce (C. sinensis) del banco de germoplasma del Campo Experimental General Terán del Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias (INIFAP) en Nuevo León, México, utilizando marcadores RAM. Todos estos marcadores moleculares producen matrices binarias de presencia o ausencia de fragmentos para analizar la diversidad genética, pero su elaboración puede ser laboriosa y propensa a errores. Alternativamente, se pueden utilizar procedimientos como metodologías de DNA barcode, incrementando la precisión de la caracterización varietal.
En plantas, se han utilizado regiones nucleares (ITS, ITS1, ITS2) y de cloroplasto (matK, rbcL, trnH-psbA, rpoB, rpoC1, trnL-trnF, psbK-psbI) (Letsiou et al., 2024). Estos marcadores se han utilizado para análisis filogenéticos con varias especies de cítricos, incluyendo a C. sinensis (Li et al., 2010; Kyndt et al., 2010; Penjor et al., 2013; Hynniewta et al., 2014; Ito et al., 2014; Jin et al., 2018; Liu et al., 2021; Abbaszadeh et al., 2023). Sin embargo, la mayor parte de investigaciones están centradas en países asiáticos. En Latinoamérica, no hay estudios que hayan utilizado la región ITS en C. sinensis para determinar su diversidad.
El objetivo de este trabajo fue evaluar la diversidad genética y realizar un análisis filogenético basados en la región ITS de seis genotipos de C. sinensis cultivados en San Luis Potosí, México.
Se recolectaron hojas jóvenes y con apariencia sana de seis árboles de naranja dulce (Cuadro 1). Las hojas se envolvieron en bolsas de papel y se colocaron en hieleras térmicas con geles refrigerantes para enviarlas al Laboratorio de Biotecnología del Campo Experimental Tecomán del INIFAP en Colima, donde fueron procesadas inmediatamente.
Se utilizó el método descrito por Bermúdez-Guzmán et al. (2016) con algunas modificaciones. El tejido vegetal (aproximadamente 200 mg) se maceró en morteros y se homogeneizó en 2 ml de buffer CTAB precalentado a 65 °C durante 60 min y se trató con RNasa A, seguido de la separación de fases con fenol:cloroformo:alcohol isoamílico (25:24:1) y precipitación del DNA con isopropanol frío y acetato de sodio. Se lavó el pellet de DNA con etanol al 70%, se secó a temperatura ambiente y se resuspendió en agua.
La cuantificación y pureza se determinó a partir de 2 µl de cada muestra, midiendo las relaciones de absorbancia 260/280 y 260/230 nm en un espectrofotómetro NanoDrop 2000 (Thermo Scientific). La integridad del DNA se verificó mediante electroforesis en gel de agarosa al 1% en buffer TAE 1X. La tinción de los ácidos nucleicos se realizó con GelRed (Biotium) y se visualizaron en un fotodocumentador (UVP).
Se utilizaron los oligonucleótidos descritos por White et al. (1990) modificados por Viglietti et al. (2019): ITS4: 5’-TCCTCYRMTTAKYGATATGC-3’ e ITS1: 5’-TCCGTWRGTGAACCWGCGG-3’. Las reacciones de PCR se realizaron en un volumen de 50 µl utilizando REDTaq® ReadyMixTM PCR Reaction Mix (Sigma Aldrich), de acuerdo con las indicaciones del fabricante.
La temperatura de alineamiento se estandarizó mediante un gradiente: 50, 51, 52, 53, 54 y 55 °C en un termociclador VeritiTM (Applied BiosystemsTM). Para todos los casos, las condiciones de amplificación fueron de 5 min a 95 °C, seguido de 35 ciclos de 1 min a 95 °C, 30 s a 50-55 °C y 1 min a 72 °C y una extensión final de 10 min a 72 °C. Los productos de PCR de entre 600-700 pb se purificaron con el kit Wizard® SV Gel and PCR Clean-Up System (Promega) y se enviaron a secuenciar al Laboratorio Nacional de Biotecnología Agrícola, Médica y Ambiental (Lanbama).
Las secuencias fueron editadas, se realizó un análisis BLAST en la base de datos de NCBI y se descargaron 28 secuencias de regiones ITS de C. sinensis para generar una base de datos local. Como grupo externo se incluyó una secuencia ITS de Ruta graveolens y se realizó un alineamiento múltiple de secuencias (AMS) utilizando el algoritmo de ClustalW.
Para el análisis de la diversidad genética, con base al AMS se generaron matrices de comparación de secuencias. La matriz de distancia fue calculada como la distancia corregida de Jukes-Cantor entre dos secuencias. Para el análisis filogenético, el AMS fue analizado con la herramienta ‘model test’. Se construyó un árbol filogenético con el método de construcción UPGMA utilizando el modelo sustitución nucleotídico GTR + G + T y un análisis Bootstrap con 1 000 réplicas. Todos los análisis bioinformáticos mencionados se llevaron a cabo con una licencia vigente del software CLC Main Workbench versión 24.0.2 (Qiagen).
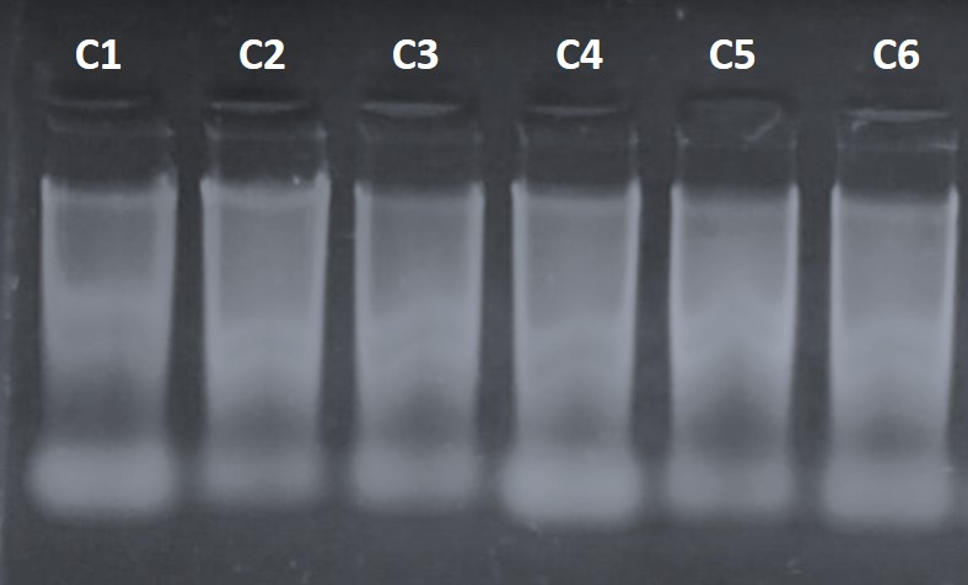
Los índices de pureza promedio fueron 1.8 (A260/280) y 1.79 (A260/230). La Figura 1 muestra la electroforesis en gel de agarosa del DNA genómico extraído, en la que se observa una degradación parcial de las muestras, probablemente debido al tiempo transcurrido desde el transporte de San Luis Potosí a Colima. No obstante, en la parte superior del gel se apreciaron bandas de DNA de alta integridad, suficientes para la amplificación por PCR de la región ITS y para la obtención de secuencias.
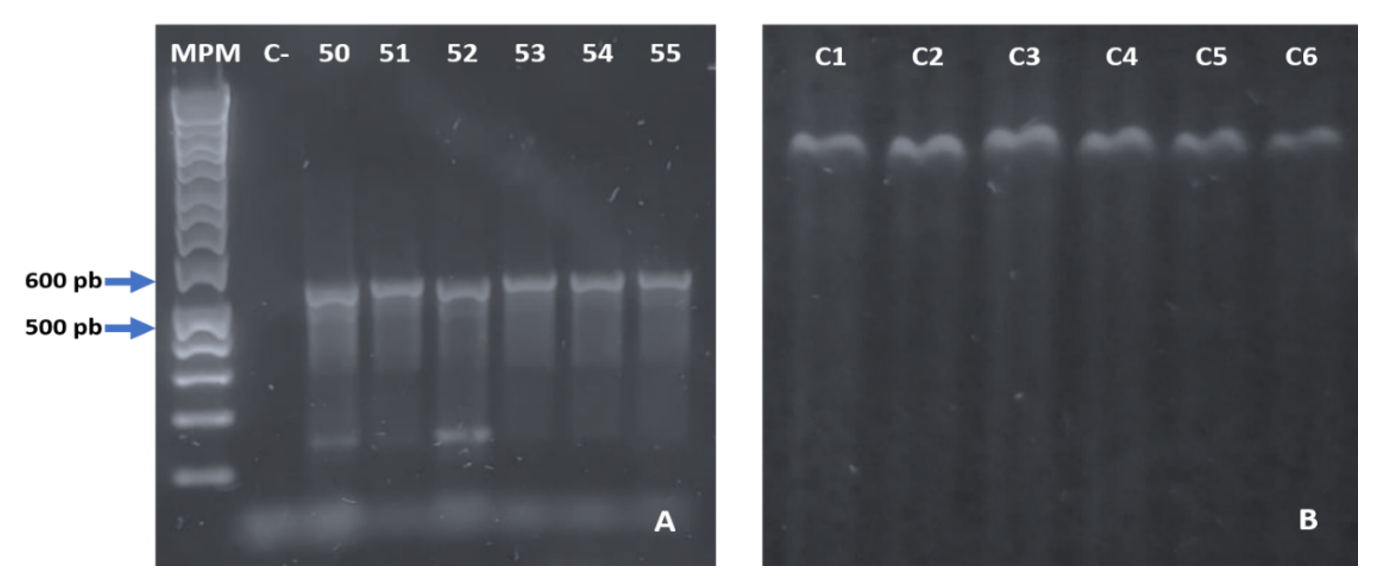
La amplificación por PCR de gradiente para evaluar el efecto de las temperaturas 50-55 °C se muestra en la Figura 2A. Las temperaturas de 53 a 55 °C mostraron bandas únicas y se seleccionó la temperatura de 54 °C para amplificar por PCR las seis muestras de árboles de naranja (Figura 2B).
En el árbol filogenético de varios genotipos de C. sinensis de diferentes regiones del mundo, se observa que los 28 genotipos se distribuyeron en cinco grupos (G1-G5) (Figura 3). Los genotipos de México formaron un grupo con otros de Corea, China y Vietnam (G1), con algunos comparte un 100% de identidad (JN681150, MG702211, MG702223, MF15502 y ON479687), constituyendo los materiales más estrechamente relacionados.
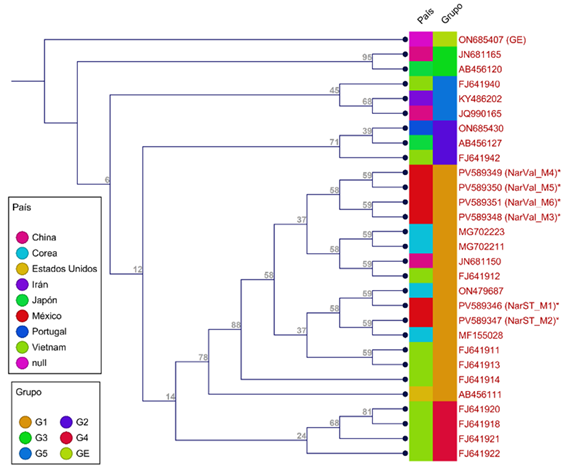
Dado que el aumento de la distancia geográfica suele incrementar la distancia genética (Abbaszadeh et al., 2023), en este estudio la variabilidad genética en la región ITS entre los seis genotipos de C. sinensis fue de 0, con secuencias homólogas, probablemente por la cercanía de los sitios de muestreo y al limitado número de muestras analizadas. Esto indica alta conservación en esta región entre las variedades Sangre de Toro y Valencia. Los genotipos de Portugal, Japón y Vietnam, que forman el grupo G5 (Figura 3), mostraron mayor distancia genética, con valores de 94.1-95.71% de identidad y coeficiente de distancia de 0.04-0.06.
La secuencia de ITS es ampliamente utilizada para identificar especies vegetales y realizar estudios filogenéticos (Alvarez y Wendel, 2003; Graper et al., 2021). En cítricos, se ha empleado con éxito en especies como C. sinensis, C. limon y C. reticulata para evaluar variabilidad genética y relaciones filogenéticas (Hynniewta et al., 2014). En este estudio, la distancia genética entre todos los genotipos de C. sinensis (28 secuencias) fue baja a moderada (0-0.07), indicando alta conservación en la región ITS, mientras que en comparación con Ruta graveolens los valores fueron de 0.24-0.29. Resultados similares fueron obtenidos por Liu et al. (2021) en 22 muestras de C. reticulata ‘Chachi’, donde la región ITS2 mostró una homología del 100%, indicando alta estabilidad genética en esta región.
Por otra parte, los genotipos de C. sinensis de Vietnam mostraron mayor variabilidad intraespecífica (distancias de 0.02-0.06) y se agruparon en G1, G2, G4 y G5. Los materiales de China estuvieron en G1, G3 y G5, mientras que los de Corea y México se conglomeraron en G1. Esta diversidad puede atribuirse a diferentes variedades, regiones climáticas y prácticas de cultivo y mejoramiento (Abbaszadeh et al., 2023; Seminara et al., 2023). A diferencia de estos resultados, Almeyda-León et al. (2022) evaluaron 33 variedades de naranjo dulce mediante RAMs, logrando un alto porcentaje de polimorfismos y diferenciando todos los materiales evaluados, lo que indica una amplia base genética en su germoplasma.
En otros trabajos utilizando marcadores RAPDs, Tuwo et al. (2023) identificaron cinco grupos con 175 genotipos de cítricos mediante un análisis de conglomerados con un coeficiente de similitud del 77%. Por otra parte, los marcadores SSR también se han utilizado para la identificación de híbridos para el mejoramiento genético en cítricos (Bermúdez-Guzmán et al., 2017; Gallego-Colonia et al., 2017; Sharafi et al., 2017; Carrillo-Medrano et al., 2018). Lo anterior sugiere que el uso de marcadores moleculares basados en perfiles electroforéticos como RAPDs, RAMs, SSR, SCoT, entre otros, siguen siendo muy útiles para caracterizar la variabilidad genética en C. sinensis y otras especies cítricas (Shahnazari et al., 2022; Abbaszadeh et al., 2023).
El uso de estos marcadores moleculares para la caracterización de plantas se remonta desde los 90’s (Williams et al., 1990; Morgante y Olivieri, 1993) pero siguen empleándose ampliamente en la actualidad ya que resultan muy versátiles porque exploran el genoma completo. En este estudio se evaluó una pequeña región de un genoma, ITS, con una longitud de poco más de 550 pb, que representa una ínfima parte del genoma completo. Por tal motivo, para hacer más robusto el análisis de diversidad es recomendable utilizar más de una región como pueden ser regiones nucleares (ITS, ITS1, ITS2) o de cloroplasto (matK, rbcL, trnH-psbA, rpoB, rpoC1, trnL-trnF, psbK-psbI) (Letsiou et al., 2024).
Por otra parte, es importante considerar que los costos de la secuenciación masiva (NGS) han disminuido significativamente, por lo que en el futuro próximo es deseable migrar a este tipo de marcadores. Con esta tecnología se puede obtener una gran cantidad de información sobre variación genética, como polimorfismos de un solo nucleótido (SNP), inserciones-deleciones (InDel), variación estructural (SV), variación en el número de copias (CNV), entre otros (Wenger et al., 2019; Ahsan et al., 2021).
La región ITS puede emplearse como herramienta exploratoria y comparativa para evaluar relaciones filogenéticas en el género Citrus; sin embargo, su resolución intraespecífica resultó ser limitada en materiales estrechamente relacionados, como fue el caso de los genotipos de C. sinensis. En este estudio, con seis muestras correspondientes a dos variedades de San Luis Potosí (SLP), se observó una variabilidad intraespecífica baja a moderada (0-0.07) entre diversos genotipos de C. sinensis de varias partes del mundo. Estas conclusiones son específicas de las muestras analizadas de la región de SLP. Las secuencias de los genotipos mostraron similitudes con accesiones asiáticas (China, Corea y Vietnam), pero se requiere ampliar la cobertura genómica para confirmar estos posibles orígenes.
Se agradece al INIFAP por el financiamiento del proyecto titulado: ‘búsqueda de mutantes naturales de cítricos con características de interés agronómico, fitosanitario e industrial en la región noreste de México’.
Almeyda-León, I. H.; Narvaéz-Rodríguez, Á. I.; Pecina-Quintero, V.; Álvarez-Ojeda, M. G. y Núñez-Colín, C. A. 2023. Caracterización y diversidad genética de cítricos del banco de germoplasma del Campo Experimental General Terán. Biotecnología y Sustentabilidad. 8(1):26-41. https://doi.org/10.57737/biotecnologiaysust.v8i1.2276.
Almeyda-León, I. H.; Pecina-Quintero, V.; Álvarez-Ojeda, M. G.; Rodríguez-Guerra, R.; Acosta-Díaz, E. y Núñez-Colín, C. A. 2022. Caracterización y diversidad genética de naranjos dulces (Citrus sinensis L.) del banco de germoplasma del Campo Experimental General Terán. Biotecnología y Sustentabilidad. 7(1):80-95. https://doi.org/10.57737/biotecnologiaysust.v7i1.1645.
Bermúdez-Guzmán, M. J.; Guzmán-Rodríguez, L. F.; García-Mariscal, K. P.; Palmeros-Suárez, P. A. y Orozco-Santos, M. 2017. Identificación de híbridos de Citrus aurantifolia×Citrus limon utilizando marcadores de secuencias simples repetidas (SSR). Revista Mexicana de Ciencias Agrícolas. 8(6):1397-1408. https://doi.org/10.29312/remexca.v8i6.309.
Gallego-Colonia, J. S.; Enríquez-Valencia, A. L.; Caicedo-Arana, Á.; Posso-Terranova, A. M. y Muñoz-Florez, J. E. 2017. Diversidad genética en patrones de cítricos mediante microsatélites amplificados al azar (RAMs). Biotecnología en el Sector Agropecuario y Agroindustrial. 15(1):85-94. https://doi.org/10.18684/BSAA(15)85-94.
Ito, T. M.; Polido, P. B.; Rampim, M. C.; Kaschuk, G. and Souza, S. G. H. 2014. Genome-wide identification and phylogenetic analysis of the AP2/ERF gene superfamily in sweet orange (Citrus sinensis). Genetics and Molecular Research: GMR. 13(3):7839-7851. https://doi.org/10.4238/2014.September.26.22.
Jin, S. B.; Lee, W. J.; Park, J. H.; Park, S. M.; Lee, D. H. and Yun, S. H. 2018. A phylogenic analysis of citrus cultivars native to jeju using chloroplast dna trnl-trnf and internal transcribed spacer region sequences. Horticultural Science and Technology. 36(4):585-597. https://doi.org/10.12972/kjhst.20180059.
Li, X.; Xie, R.; Lu, Z. and Zhou, Z. 2010. The origin of cultivated citrus as inferred from internal transcribed spacer and chloroplast DNA sequence and amplified fragment length polymorphism fingerprints. Journal of the American Society for Horticultural Science. 135(4):341-350. https://doi.org/10.21273/JASHS.135.4.341.
Liu, M.; Wang, K.; Chen, B.; Cai, Y; Li, C.; Yang, W.; Wei, M. and Zheng, G. 2021. Intraspecific DNA barcoding and variation analysis for Citri reticulatae pericarpium of Citrus reticulata ‘Chachi’. Evidence-based Complementary and Alternative Medicine. eCAM, 2021, 2609935: 1-7 pp. https://doi.org/10.1155/2021/2609935.
Wenger, A. M.; Peluso, P.; Rowell, W. J.; Chang, P. C.; Hall, R. J.; Concepcion, G. T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N. D.; Töpfer, A.; Chin, C. S.; Alonge, M.; Mahmoud, M.; Qian, Y.; Phillippy, A. M.; Schatz, M. C.; Myers, G.; Pristo, M. A. and Hunkapiller, M. W. 2019. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. Preprint, bioRxiv, 519025. https://doi.org/10.1101/519025.
White, T.; Bruns, T.; Lee, S.; Taylor, J.; Innis, M.; Gelfand, D. and Sninsky, J. J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols: A Guide to Methods and Applications. Academic Press. San Diego, California, USA. 315-322 pp. https://doi.org/10.1016/b978-0-12-372180-8.50042-1.